MMPBSA with membrane proteins¶
Info
This example can be found in the examples/Protein_membrane directory in the repository folder. If you didn't use gmx_MMPBSA_test before, use downgit to download the specific folder from gmx_MMPBSA GitHub repository.
Requirements¶
Danger
The ligand mol2 file must be the Antechamber output.
In this case, gmx_MMPBSA requires:
| Input File required | Required | Type | Description |
|---|---|---|---|
| Input parameters file | in | Input file containing all the specifications regarding the type of calculation that is going to be performed | |
| The MD Structure+mass(db) file | tpr pdb | Structure file containing the system coordinates | |
| An index file | ndx | file containing the receptor and ligand in separated groups | |
| Receptor and ligand group | integers | Receptor and ligand group numbers in the index file | |
| A trajectory file | xtc pdb trr | Final GROMACS MD trajectory, fitted and with no pbc. | |
| Ligand parameters file | mol2 | The Antechamber output mol2 file of ligand parametrization | |
| A topology file (not included) | top | GROMACS topology file (The * .itp files defined in the topology must be in the same folder | |
| A Reference Structure file | pdb | Complex reference structure file (without hydrogens) with the desired assignment of chain ID and residue numbers |
-> Must be defined -- -> Optional, but recommended -- -> Optional
Remember
When a topology file is defined, the ligand mol2 file is not needed. The ligand mol2 file only required when
gmx_MMPBSA build the amber topology from a structure
See a detailed list of all the flags in gmx_MMPBSA command line here
Command-line¶
That being said, once you are in the folder containing all files, the command-line will be as follows:
gmx_MMPBSA -O -i mmpbsa.in -cs com.pdb -ci index.ndx -cg 1 13 -ct com_traj.pdb -lm ligand.mol2 -o FINAL_RESULTS_MMPBSA.dat -eo FINAL_RESULTS_MMPBSA.csv
mpirun -np 2 gmx_MMPBSA -O -i mmpbsa.in -cs com.pdb -ci index.ndx -cg 1 13 -ct com_traj.pdb -lm ligand.mol2 -o FINAL_RESULTS_MMPBSA.dat -eo FINAL_RESULTS_MMPBSA.csv
gmx_MMPBSA_test -t 6
where the mmpbsa.in input file, is a text file containing the following lines:
Keep in mind
See a detailed list of all the options in gmx_MMPBSA input file here as well as several examples. These examples are meant only to show that gmx_MMPBSA works. It is recommended to go over these variables, even the ones that are not included in this input file but are available for the calculation that it's performed and see the values they can take (check the input file section). This will allow you to tackle a number of potential problems or simply use fancier approximations in your calculations.
Considerations¶
In this case, a single trajectory (ST) approximation is followed, which means the receptor and ligand structures and trajectories will be obtained from that of the complex. To do so, an MD Structure+mass(db) file (com.pdb), an index file (index.ndx), a trajectory file (com_traj.pdb), and both the receptor and ligand group numbers in the index file (1 13) are needed. A ligand .mol2 file is also needed for generating the ligand topology. The mmpbsa. in input file will contain all the parameters needed for the MM/PB(GB)SA calculation. Of note, special parameters for MMPBSA with membrane proteins have been included.
Comments on parameters for implicit membranes
The inclusion of an implicit membrane region in implicit solvation calculations is enabled by setting memopt to 1 (default value is 0, for off). The membrane will extend the solute dielectric region to include a slab-like planar region of uniform dielectric constant running parallel to the xy plane. The dielectric constant can be controlled using emem. We set the membrane interior dielectric constant to a value of 7.0 in this example. The value of emem should always be set to a value greater than or equal to indi (solute dielectric constant, 4 in this example) and less than exdi (solvent dielectric constant, 80.0 default).
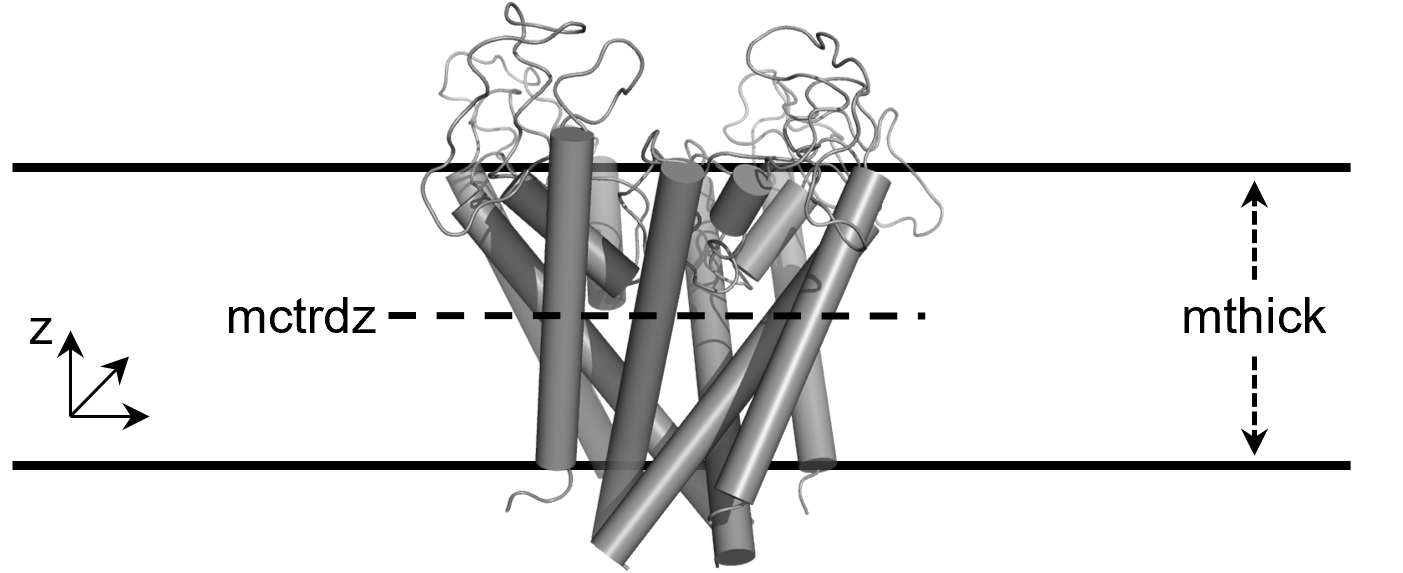
The thickness is controlled by the mthick option (36.086 Å in this case). The center of the membrane region is controlled with mctrdz and in this case the membrane region will be centered at -10.383 Å down of the center of the protein. If calculations are performed on a protein with a solvent-filled channel region, this region would be identified automatically by setting poretype=1.
When using the implicit membrane model, the default sasopt=0, i.e. the classical solvent excluded surface, is recommended due to its better numerical behavior. When running with the default options, the program will compute solvent excluded surfaces both with the water probe (prbrad=1.40 by default) and the membrane probe (mprob=2.70 by default). This setting was found to be consistent with the explicit solvent MD simulations.
It is also suggested that periodic boundary conditions be used to avoid unphysical edge effects. This is supported in all linear solvers. In the following, Geometric multigrid is chosen (solvopt=2) with ipb=1 and bcopt=10. The linit=1000 should work fine, but take into account that working with linear and periodic boundary conditions could require more iterations.
In addition, eneopt needs to be set to 1 because the charge-view method (eneopt = 2) is not supported for this application. When eneopt=1, the total electrostatic energy and forces will be computed with the particle-particle particle-mesh (P3M) procedure outlined in Lu and Luo.[8] In doing so, energy term EPB in the output file is set to zero, while EEL term includes both the reaction field energy (EPB) and the Coulombic energy (EEL). The van der Waals energy is computed along with the particle-particle portion of the Coulombic energy. This option requires a nonzero CUTNB (in this case, cutnb=8.0). It's noteworthy mentioning that ΔGGAS and ΔGSOLV as reported are no longer properly decomposed. Since EPB and EEL are combined into the "gas phase" term, the gas and solvation terms can't be separated. Nevertheless, the total ΔTOTAL should be perfectly fine, since everything is sum up together in the end.
Danger
Note that a smaller fillratio=1.25 is used compared to the defult one (4.0). The use of a periodic boundary also allowed a somewhat small fill ratio (i.e., the ratio of the finite-difference box dimension over the solute dimension) of 1.25 to be used in these calculations (ref). Be cautious when changing this parameter as its increase may lead to a considerable RAM usage (specially when running the program in parralel). Just for your information, using the default fillratio=4.0 in this relatively small system requieres as much as ~30GB per thread  and more time to finish the calculation.
and more time to finish the calculation.
A plain text output file with all the statistics (default: FINAL_RESULTS_MMPBSA.dat) and a CSV-format output file containing all energy terms for every frame in every calculation will be saved. The file name in '-eo' flag will be forced to end in [.csv] (FINAL_RESULTS_MMPBSA.csv in this case). This file is only written when specified on the command-line.
Note
Once the calculation is done, the results can be analyzed in gmx_MMPBSA_ana (if -nogui flag was not used in the command-line). Please, check the gmx_MMPBSA_ana section for more information
Created: 2023-06-24